DUB crosstalk in colorectal cancer
Colorectal cancer is the third most common cancer type (second most common in women, third most common in men) in the world, with 1.4 million new cases diagnosed in 2012. 95% of colorectal cancers are classified as adenocarcinomas. Other lesions are mucinous carcinoma and adenosquamous carcinomas.
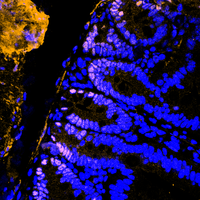
The ubiquitin-proteasome system tightly regulates the protein stability of the WNT effector molecule CTNNB1 (beta-Catenin). Several E3 ligases are involved in its regulation, ranging from decreasing its stability to enhancing its function as a transcriptional co-factor, whereas the contribution of DUBs remains less explored. In the intestine, CTNNB1 protein abundance is limited to the intestinal stem cell compartment via a WNT ligand gradient, generated by paneth cells and fibroblasts. Soluble WNT ligands inactivate the WNT destruction complex by recruiting it to the membrane. This complex is composed of APC, CK1-alpha, GSK3-beta, AXIN and CTNNB1. Upon gradient-loss, the WNT destruction complex binds to CTNNB1, is phosphorylated by CK1-alpha and GSK3-beta, followed by SCF-beta-TrcP-dependent proteasomal degradation. This process is crucial to maintain complex tissue homeostasis and to prevent oncogenic transformation, as CTNNB1 is a potent proto-oncogene driving de-differentiation and hyperproliferation.
Colorectal cancer (CRC) is among the leading causes of death worldwide in 2020, accounting for one in six deaths, according to WHO. In the U.S. Cancer Statistics 2021, colorectal cancer is the third most common cancer in both men and women, with an estimated 8% new cases and an estimated 8% to 9% deaths in 2021. Interestingly, the likelihood (in %) of developing invasive colorectal cancer increases with age. The reasons for the age differences are likely due to differential exposure to environmental risk factors and mutational burden that accumulates over time. It is widely recognized that factors such as smoking, alcohol, diet, and obesity increase the risk of colorectal cancer. In addition to these environmental risk factors, there are also known inherited forms of colorectal cancer characterized by specific mutations. Although the majority of patients have an overall good survival rate after diagnosis, only 10% of patients diagnosed at stage 4 survive longer than 5 years. These poor prognoses for late stage patients underscore the need to uncover the molecular mechanisms underlying sporadic and hereditary colorectal cancers.
In colorectal cancer (CRC), the tumour suppressor APC is commonly lost (~76%), leading to stabilization and accumulation of CTNNB1. This constitutes one of the main tumor-initiating genetic event. During the initiation of transformation, enzymes of the UPS system are altered in their abundance or activity. Tumor cells rely on the ubiquitin system to initiate and maintain oncogenic capacity of diverse human tumors. Genes encoding E3 ligases and deubiquitylases (DUBs) are often amplified, mutated, or lost in tumors. This results in dysregulation of proteasome-dependent degradation of key proteins in oncogenic reprogramming (e.g. β-Catenin, or c-MYC) or leads to the adaptation of the tumor cell machinery to increased proliferation, metabolic changes and stress management. In such a way, cancer cells are dependent on regulators of the ubiquitin system (E3 ligases or DUBs) which may therefore be useful as potential anti-cancer therapy targets.
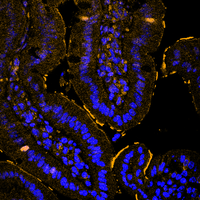
In our laboratory, we are interested in recapitulating genetic alterations that lead to sporadic and hereditary colorectal cancer using the CRISPR/Cas9 system and our ex vivo colon organoids. Our goal is to understand the development of colon cancer by recapitulating the sequence from adenoma to carcinoma and using these models to identify and validate novel important factors in the development and maintenance of colon cancer. In particular, we are investigating how the UPS is altered at various stages of CRC onset and progression, and which enzymes can be exploited for therapeutic intervention.